
Britse onderzoekers hebben de eerste evolutionaire paden in kaart gebracht waarlangs het coronavirus zich verspreidde van Wuhan naar Europa en Noord-Amerika. Het onderzoek laat zien dat er in deze eerste fase drie verschillende clusters van virusvarianten ontstonden.
De onderzoekers analyseerden de virusgenomen van 160 coronapatiënten. Het ging om mensen die in het begin van de pandemie – tussen december 2019 en begin maart 2020 – geïnfecteerd waren. Door naar mutaties in het virus te kijken, onderscheidden ze verschillende afstammingslijnen. Dit leverde een webachtig netwerkpatroon op met drie onderscheidbare clusters.

Seksuoloog ontwikkelt nieuwe erectiemeter: 'Mannen schrijven zichzelf te snel af'
De manier om erectieproblemen te onderzoeken is pijnlijk en verouderd. Evelien Trip ontwikkelde een nieuwe, comfortabele erectiemeter.
Het nieuwe coronavirus is een RNA-virus. Net als andere virussen verspreidt het zich door cellen van zijn gastheer ertoe aan te zetten zijn genetische materiaal te kopiëren. Die kopieën komen vrij en kunnen andere cellen (en andere gastheren) infecteren.
Dit kopiëren gebeurt alleen nogal slordig. Daardoor ontstaan er foutjes, ofwel mutaties, en komt het genetische materiaal van de kopieën er net wat anders uit te zien. Via die foutjes kun je herleiden welke virussen meer en minder aan elkaar verwant zijn. Dochtervirussen zullen veel op hun moedervirus lijken en iets minder op hun oma of tante.
Door veel virusgenomen te vergelijken, kun je dus een stamboom opstellen van de ontwikkeling van het virus. Zo kun je – in theorie – ook de eerste coronavirus-voorouder opsporen die een mens infecteerde.
‘Er zijn te veel snelle mutaties om de covid-19-stamboom netjes te herleiden’, zegt geneticus Peter Forster van de University of Cambridge. ‘Daarom gebruiken we een wiskundig netwerkalgoritme om alle plausibele stambomen tegelijkertijd te visualiseren.’ Deze techniek is al eerder ingezet om de bewegingen van prehistorische mensenpopulaties over de wereld in kaart te brengen aan de hand van gevonden DNA.
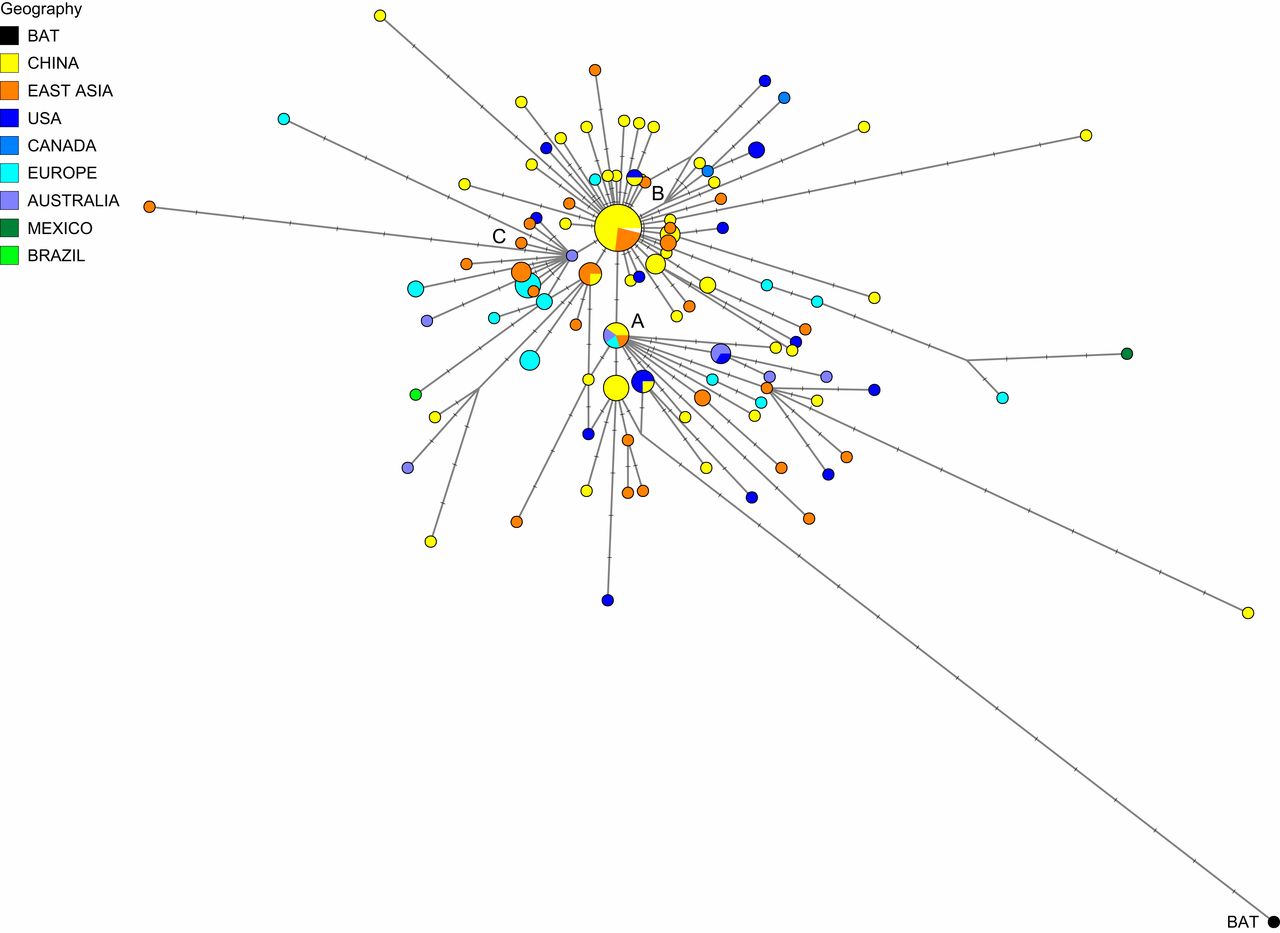
Type A, B en C
Deze netwerktechniek bracht drie verschillende clusters van nauw verwante virusafstammingslijnen in beeld: type A, B en C. Om te achterhalen welke variant het dichtst bij de eerste menselijke infectie zit, vergeleken de onderzoekers het genetische materiaal van de drie clusters met dat van het coronavirus van een vleermuis. Hieruit bleek dat variant A het meest verwant is aan het coronavirus dat voorkomt in vleermuizen (en schubdieren). A staat daarmee waarschijnlijk aan het begin van de uitbraak.
Een aftakking van type A muteerde en veranderde in type B. Deze variant besmette uiteindelijk meer mensen in Wuhan dan A. Ook in de rest van China en Oost-Azië domineerde B. Variant B produceerde vervolgens C, die vooral teruggevonden wordt in delen van Europa, Singapore, Hongkong, Taiwan en Brazilië. Op het vasteland van China werd C niet teruggevonden.
Deze drie clusters werden gevonden in virusgenomen die tot begin maart verzameld waren. Volgens Forster is het waarschijnlijk dat het virus sindsdien verder gemuteerd is waardoor er nu meer dan drie clusters te onderscheiden zijn. Of die virustypen al dusdanig verschillen dat immuniteit voor het ene niet gelijk staat aan immuniteit voor het andere, is nog niet te zeggen.
Meer virusgenomen
Inmiddels heeft de onderzoekersgroep, voortbouwend op dit werk, de analyse uitgebreid tot ruim duizend virale genomen. Volgen Forster lijkt dit vervolgonderzoek erop te wijzen dat de eerste infectie en verspreiding onder mensen van covid-19 plaatsvond tussen half september en begin december.
Dankzij onderzoekers en klinieken wereldwijd die genoomdata vrijgeven, blijft de database met virusgenomen groeien. ‘Deze netwerkanalyse kan in de toekomst mogelijk helpen om covid-19-infectiebronnen te identificeren. Dan kunnen die vervolgens in quarantaine worden geplaatst om verdere verspreiding van de ziekte te voorkomen’, zegt Forster.